
Synopsis
This is an interventional, randomised, double-blind, placebo-controlled, two-armed, parallel-group, multicentre, multinational clinical study. The study is primarily intended to assess the cardiovascular (CV) safety and potential CV benefits of CagriSema 2.4 mg/2.4 mg versus placebo, both administered subcutaneously (s.c.) once weekly and both added to standard of care in individuals with established cardiovascular disease (CVD), with or without comorbidities such as obesity, overweight, T2D and CKD. The study design will also allow for evaluation of neoplasm safety, due to the treatment duration.
Rationale
Cagrilintide is a new active substance and considered a ‘first in class’ medicinal product in the EU, therefore, a dedicated CV outcome study to evaluate the CV safety profile is required by the EMA. The study has been designed to address requirements contained in the EMA reflection paper2 for evaluation of the CV safety profile of new medicinal products that are intended for long-term treatment of certain CV and metabolic diseases.
The primary purpose of this study is to investigate the long-term CV safety of CagriSema as fixed dose combination of cagrilintide and semaglutide in participants with established CVD. Secondly, the study will investigate the efficacy of CagriSema on CV risk reduction.
Objectives, endpoints and estimand:
| Objectives | Endpoints | ||||||
|---|---|---|---|---|---|---|---|
| Primary | Primary endpoint | ||||||
| To confirm non-inferiority of CagriSema 2.4 mg/2.4 mg versus placebo with respect to time to first major adverse cardiovascular event (MACE) |
|
||||||
| Secondary | |||||||
| To confirm superiority of CagriSema 2.4 mg/2.4 mg versus placebo with respect to time to first MACE |
aMaximum treatment duration is dependent on event rates and is estimated to be approximately 242 weeks including a 7-week follow-up period.
bBased on event adjudication committee (EAC)-confirmed events; cincluding undetermined cause of death; dacute myocardial infarction only; eincluding ischaemic, haemorrhagic and undetermined stroke.
Primary estimand
The primary estimand addresses the hazard ratio for first occurrence of MACE for CagriSema 2.4 mg/2.4 mg versus placebo in participants with established CVD with or without comorbidities such as obesity, overweight, T2D or CKD, regardless of adherence to treatment and changes to dose, where both CagriSema and placebo are added to standard of care.
The primary estimand will be used for both primary and confirmatory secondary comparisons.
Overall design:
The study consists of a screening period to assess the participant’s eligibility, an up to 235-week treatment period (including a 16-week dose escalation period) and a 7-week follow-up period. The total study duration for each participant will be up to approximately 245 weeks (up to 3 weeks screening, up to 235 weeks treatment and 7 weeks follow-up). The study is event driven; therefore, end of study will be scheduled according to projected study closure. Study duration is expected to be up to approximately 4.5 years following randomisation of the first participant.
Study intervention and duration:
The following IMPs will be supplied by Novo Nordisk:
| Treatment arm | CagriSema | Placebo |
|---|---|---|
| Intervention name | A: Cagrilintide + semaglutide | B: Placebo cagrilintide + placebo semaglutide |
| IMPs | A1: Cagrilintide B 1.0 mg/mL and semaglutide I 0.5 mg/mL A2: Cagrilintide B 2.0 mg/mL and semaglutide I 1.0 mg/mL A3: Cagrilintide B 4.0 mg/mL and semaglutide I 2.0 mg/mL A4: Cagrilintide B 6.8 mg/mL and semaglutide I 3.4 mg/mL A5: Cagrilintide B 9.6 mg/mL and semaglutide I 4.8 mg/mL |
B: Placebo cagrilintide B and placebo semaglutide I |
| Route of administration | subcutaneous | |
| Packaging and labelling | The DV3384 pen-injector is a dual chamber single-dose device. The DV3384 pen-injector administers the two products as a single dose. | |
Number of participants:
Approximately 7000 eligible participants will be assigned to randomised treatment in a 1:1 manner to CagriSema or placebo both added to standard of care.
Participant characteristics:
Participants eligible for this study are male or female, age ≥ 55 years with established CVD, with or without comorbidities such as obesity, overweight, T2D and CKD. The study will include approximately 80% of total participants with T2D and approximately 30% of total participants with stage 3 to 4 CKD (all combinations allowed).
Key inclusion criteria
- Male or female
- Age above or equal to 55 years at the time of signing informed consent.
- Body mass index (BMI) ≥ 25.0 kg/m2
- Established CVD as evidenced by at least one of the following:
- Prior myocardial infarction
- Prior stroke (ischemic or haemorrhagic stroke)
- Symptomatic peripheral arterial disease (PAD) defined as at least one of the following:
- Intermittent claudication with an ankle-brachial index (ABI) < 0.85 at rest
- Intermittent claudication with a ≥ 50% stenosis in a lower extremity peripheral artery
documented by X-ray angiography, MR angiography, CT angiography or Doppler ultrasound - Prior revascularization procedure of a lower extremity peripheral artery
- Lower extremity amputation at or above ankle due to atherosclerotic disease (excluding e.g.,
trauma or osteomyelitis)
For participants with T2D at screening the following inclusion criteria also apply
- Diagnosed with type 2 diabetes mellitus (T2D) ≥ 180 days before screening
- HbA 1c 6.5%-10% (48-86 mmol/mol) (both inclusive), as measured by central laboratory at
screening. - Treatment with either:
- Lifestyle intervention alone
- 1-3 marketed oral antidiabetic drugs (OADs) (metformin, o-glucosidase inhibitors (AGI), glinides,
sodium-glucose co-transporter 2 inhibitor (SGLT2i), DPP4- inhibitors,
thiazolidinediones, or sulphonylureas (SU) as a single agent or in combination) according to
local label - Basal insulin alone or in combination with up to two marketed OADs (refer to b. above), all
according to local label
Key exclusion criteria
- Myocardial infarction, stroke, hospitalization for unstable angina pectoris or transient ischaemic attack within 60 days before screening
- Planned coronary, carotid or peripheral artery revascularisation known on the day of screening
- Heart failure classified as being in New York Heart Association (NYHA) Class IV at screening
- Treatment with any GLP-1 RA or a medication with GLP-1 activity within 90 days before screening
- End stage renal disease defined as eGFR < 15 mL/min/1.73 m2, as measured by the central laboratory at screening
- Chronic or intermittent haemodialysis or peritoneal dialysis
Data monitoring committee: Yes

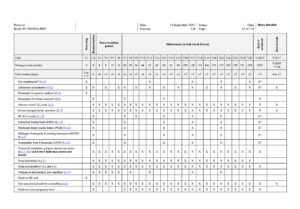

- Prior to end of study visit, 2 hours of fasting is required
- Period between screening and randomisation should be from 16 to 21 days for women of childbearing potential. For men and women of non-childbearing potential the period should be from 7 to 21 days
- Demography consists of date of birth, sex, ethnicity, and race (according to local regulation). Race and ethnicity must be self-reported by the participant
- Only for female participants
- Only for women of childbearing potential. The pregnancy test at V1 is a serum pregnancy test, all other pregnancy tests are urine pregnancy tests. A urine pregnancy test is required every 4 weeks after randomisation. After week 16, remote contacts are required every 4 weeks between visits in order to confirm the results of a urine pregnancy test. The participants will be provided with urine pregnancy tests for remote testing. The participants should be instructed by site staff in correct use of the tests. In addition to the planned assessments, urine pregnancy test should be performed at any time during the study if a menstrual period is missed, or if pregnancy is otherwise suspected.
- Only for participants with type 2 diabetes at screening
- Only for participants who consented to providing these samples
- Only serious adverse events and selected other adverse events are required to be reported i) Site to collect NRS forms filled out by participants 7 consecutive days prior to visit
- Includes questionnaire and foot exam
- Pen injector training should be repeated as needed throughout the study to ensure correct use of the pen-injector
- If deemed helpful by the investigator, participants may be provided with a BG meter including auxiliaries as well as instructions for use. m) If done within the past 5 weeks, assessment can be omitted.
- Urine collection cup with lid should be given to participant prior to end of treatment visit.
- End of treatment and end of study visits will be scheduled according to study completion.
- Site to distribute NRS forms to participants prior to end of treatment visit.
