An Operationally Seamless Phase 2/3 Randomized, Open-label, Multicenter, Active-Controlled Study to Evaluate the Safety and Efficacy of Bictegravir/Lenacapavir Versus Stable Baseline Regimen in Virologically Suppressed People With HIV-1 on Stable Complex Treatment Regimens
Study Centers Planned: Approximately 110 centers globally.
Objectives and Endpoints:
| Primary Objectives | Primary Endpoints |
|---|---|
Phase 2:
Phase 3:
|
Phase 2:
Phase 3:
|
| Secondary Objectives | Secondary Endpoints |
Phase 2:
Phase 3:
|
Phase 2:
Phase 3:
|
| Exploratory Objectives | Exploratory Endpoints |
Phase 3
|
Phase 3
|
Study Design:
This study is an operationally seamless Phase 2/3 randomized, open-label, multicenter, active-controlled study to evaluate the safety and efficacy of BIC/LEN versus SBR in virologically suppressed PWH on a stable complex treatment regimen for at least 6 months prior to screening. In the Phase 2 portion, single agent BIC and LEN tablets (BIC plus LEN) will be coadministered to determine the optimal doses to be used in subsequent portions of the study. In the Phase 3 portion of the study, a single BIC/LEN FDC tablet will be administered.
Phase 2:
A randomized, open-label, multicenter, active-controlled study to evaluate the safety and efficacy of BIC plus LEN versus SBR in PWH who are virologically suppressed (HIV-1 RNA < 50 copies/mL) on a stable complex treatment regimen for at least 6 months prior to screening.
Participants will be randomized in a 2:2:1 ratio to one of the following 3 treatment groups:
- Treatment Group 1 (n = 50): Participants will switch from their SBR to a regimen of BIC 75 mg plus LEN 25 mg administered orally, without regard to food. Participants will receive a 2-day PK oral loading dose regimen of LEN 600 mg (2 300 mg LEN on Day 1 and 2 300 mg LEN on Day 2), in addition to the daily doses of BIC 75 mg plus LEN 25 mg starting on Day 1.
- Treatment Group 2 (n = 50): Participants will switch from their SBR to a regimen of BIC 75 mg plus LEN 50 mg administered orally, without regard to food. Participants will receive a 2-day PK oral loading dose regimen of LEN 600 mg (2 300 mg LEN on Day 1 and 2 300 mg LEN on Day 2), in addition to the daily doses of BIC 75 mg plus LEN 50 mg starting on Day 1.
- Treatment Group 3 (n = 25): Participants will continue with their SBR as defined above.
Phase 3:
A randomized, open-label, multicenter, active-controlled study to evaluate the safety and efficacy of FDC tablets containing either BIC 50 to 75 mg/LEN 25 mg or BIC 50 to
75 mg/LEN 50 mg (dose selection per the Week 24 data from Phase 2 portion of the current study and Phase 1 relative bioavailability (rBA) Study GS-US-621-6292) versus SBR in PWH who are virologically suppressed (HIV-1 RNA < 50 copies/mL) on a SBR for at least 6 months prior to screening.
Participants will be randomized in a 2:1 ratio to one of the following 2 treatment groups:
- Treatment Group 1 (n = 364): Participants will switch from their SBR to a regimen of BIC 50 to 75 mg/LEN 25 mg or 50 mg (final dose to be decided) administered orally, without regard to food. Participants will receive a 2-day PK oral loading dose regimen of LEN 600 mg (2 300 mg LEN on Day 1 and 2 300 mg LEN on Day 2), in addition to the daily doses of BIC/LEN FDC starting on Day 1.
- Treatment Group 2 (n = 182): Participants will continue with their SBR as defined above.
Number of Participants Planned:
Approximately 671 participants total: 125 participants in Phase 2 (50 each in Treatment Groups 1 and 2, and 25 in Treatment Group 3) and 546 participants in Phase 3 (364 in Treatment Group 1 and 182 in Treatment Group 2).
Target Population: PWH (≥ 18 years of age), virologically suppressed for at least 6 months prior to screening on a stable complex treatment regimen.
Duration of Intervention:
Phase 2:
Participants will be treated for at least 24 weeks during the Randomized Period.
After Week 24, participants will continue to take their study drug (BIC plus LEN or SBR) and attend visits every 12 weeks. Once the last participant completes Week 24 and the dose is selected for the BIC/LEN FDC, all participants will attend the end of randomized treatment (ERT) visit and will be given the option to participate in an Extension Period to receive BIC/LEN FDC at the selected dose, until this product becomes commercially available or accessible to participants through an access program, or until Gilead Sciences (Gilead) elects to discontinue the study, whichever occurs first.
Phase 3
Participants will be treated for at least 48 weeks during the Randomized Period.
After Week 48, participants will continue to take their study drug (BIC/LEN FDC at the selected dose or SBR) and attend visits every 12 weeks. Once the last participant completes the Week 48 visit and the safety and efficacy of BIC/LEN compared to the baseline regimen are demonstrated by the Week 48 analysis, all participants will attend the ERT visit and will be given the option to receive the BIC/LEN FDC tablet at the selected dose in an Extension Period until the product becomes commercially available or accessible to participants through an open access program, or until Gilead elects to discontinue the study, whichever occurs first.
Diagnosis and Main Eligibility Criteria:
Medically stable PWH who meet the following criteria:
- Documented plasma HIV-1 RNA levels < 50 copies/mL during treatment with the baseline regimen for a minimum period of 6 months prior to the screening visit.
- Plasma HIV-1 RNA levels < 50 copies/mL at screening.
- Currently receiving a complex antiretroviral (ARV) regimen due to previous viral resistance, or intolerance, or contraindication to existing single-tablet regimens. The criteria to define a complex regimen in this study are as follows:
- A regimen containing a boosted protease inhibitor or a nonnucleos(t)ide reverse transcriptase inhibitor plus at least 1 other third agent (ie, an agent from a class other than NRTIs) (eg, bictegravir/emtricitabine/tenofovir alafenamide [coformulated; Biktarvy®; BVY] + darunavir/cobicistat, BVY + etravirine), or
- A regimen of ≥ 2 pills/day, or a regimen requiring dosing more than once daily, or
- A regimen containing parenteral agent(s) (excluding a complete long-acting injectable regimen, such as intramuscular cabotegravir plus rilpivirine) as well as oral agents.
- No documented or suspected resistance to BIC (T66A/I/K, E92G/Q, G118R, F121Y, Y143C/H/R, S147G, Q148H/K/R, N155H/S, or R263K in the integrase gene).
- Estimated glomerular filtration rate ≥ 15 mL/min according to the Cockcroft-Gault formula for creatinine clearance who are not on renal replacement therapy.
- No prior use of, or exposure to, LEN.
- No chronic hepatitis B virus (HBV) infection, as determined by either:
- Positive HBV surface antigen and negative HBV surface antibody, regardless of HBV core antibody status, at the screening visit
- Positive HBV core antibody and negative HBV surface antibody, regardless of HBV surface antigen status, at the screening visit
Study Procedures/Frequency:
Phase 2:
Randomization Period: Following the Day 1 visit, participants will be required to return
for study visits at Day 2 (oral loading dose for Treatment Groups 1 and 2), Weeks 4, 12, and 24. According to the participant preference, an optional visit at Week 16 may be scheduled for blood collection for the optional intensive PK substudy to facilitate study procedures scheduled for visit Week 24. Participants will be followed every 12 weeks after the Week 24 visit until the ERT visit.
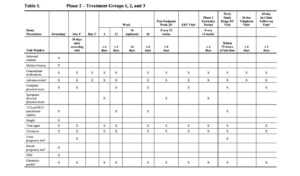
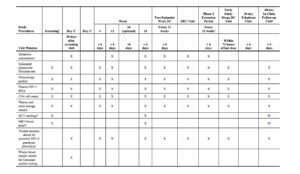
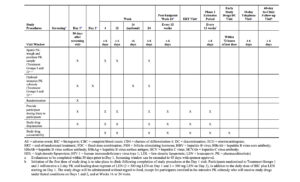
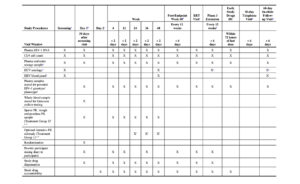
The timings of efficacy and safety assessments for the Phase 2 Randomized Period are detailed in Table 1.
Sparse PK blood samples will be collected at on-treatment study visits for all participants in Treatment Groups 1 and 2 at the time points specified in Table 1. An intensive PK substudy will be performed in a subset of participants in Treatment Groups 1 and 2 at selected study sites (Table 1).
Extension Period: Participants from Treatment Groups 1, 2, and 3 who complete the ERT visit will be given the option to enter an Extension Period. Participants who received BIC plus LEN tablets (ie, Treatment Groups 1 and 2) during the Randomized Period will continue treatment at the selected dose of BIC/LEN FDC tablet without any oral loading doses of LEN. Participants who received their SBR (ie, Treatment Group 3) during the Randomized Period will receive a 2-day PK oral loading dose regimen of LEN 600 mg (2 300 mg LEN on Day 1 and 2 300 mg LEN on Day 2), in addition to the daily selected dose of BIC/LEN FDC tablet starting on Day 1 of the Extension Period.
All participants enrolled in the Extension Period will return for study visits every 12 weeks and will have a telephone visit at 30 days and a clinic visit at 60 days after the end of the Extension Period.
Participants who complete the study through the ERT visit of the Randomized Period and do not wish to participate in the Extension Period will have a telephone visit at 30 days and a clinic visit at 60 days after the ERT visit, and will be considered to have completed the Phase 2 portion of the study.
The timings of efficacy and safety assessments for the Phase 2 Extension Period are detailed in Table 1.
Phase 3:
Randomization Period: Following the Day 1 visit, participants will be required to return for study visits at Weeks 4, 12, 24, 36, and 48. Participants will be followed every 12 weeks after the Week 48 visit until the ERT visit.
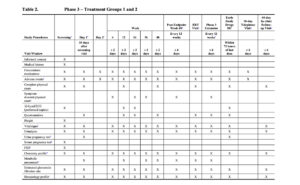
The timings of efficacy and safety assessments for the Phase 3 Randomized Period are detailed in Table 2.
Sparse PK blood samples will be collected at on-treatment study visits for all participants in Treatment Group 1 at the time points specified in Table 2. An intensive PK substudy will be performed in a subset of participants in Treatment Group 1 at selected study sites (Table 2).
Extension Period: Participants from Treatment Groups 1 and 2 who complete the ERT visit will be given the option of receiving continued treatment with the BIC/LEN FDC tablet every 12 weeks. Participants who received the BIC/LEN FDC tablet (ie, Treatment Group 1) during the Randomized Period will continue treatment at the selected dose of BIC/LEN FDC tablet without any oral loading doses of LEN. Participants from Treatment Group 2 who enroll in the Extension Period will receive a 2-day PK oral loading dose regimen of LEN 600 mg
(2 300 mg LEN on Day 1 and 2 300 mg LEN on Day 2), in addition to the daily doses of BIC/LEN FDC starting on Day 1 of the Extension Period.
All participants enrolled in the Extension Period will return for study visits every 12 weeks and will have a telephone visit at 30 days and a clinic visit at 60 days after the end of the Extension Period.
Participants who complete the study through the ERT visit of the Randomized Period and do not wish to participate in the Extension Period will have a telephone visit at 30 days and a clinic visit at 60 days after the ERT visit, and will be considered to have completed the Phase 3 portion of the study.
The timings for efficacy and safety assessments for the Phase 3 Extension Period are detailed in Table 2.
Test Product, Dose, and Mode of Administration:
Phase 2:
Bictegravir 75 mg plus LEN 25 mg, once daily, orally (Treatment Group 1); BIC 75 mg plus LEN 50 mg, once daily, orally (Treatment Group 2). Participants randomized to Treatment Groups 1 and 2 will receive a 2-day PK oral loading dose regimen of LEN (2 300 mg LEN on Day 1 and 2 300 mg LEN on Day 2), in addition to the daily doses of BIC plus LEN starting on Day 1. The study drugs will be administered without regard to food, except for participants enrolled in intensive PK substudy who will receive study drugs under fasted conditions on Days 1 and 2, and at Weeks 16 or 24 visits.
Phase 3:
Bictegravir 50 to 75 mg/LEN 25 or 50 mg (final dose to be selected) FDC
(Treatment Group 1). Participants randomized to Treatment Group 1 will receive a 2-day
PK oral loading dose regimen of LEN (2 300 mg LEN on Day 1 and 2 300 mg LEN on Day 2), in addition to the daily dose of BIC/LEN FDC tablet starting on Day 1. The study drug will be administered without regard to food.
Reference Therapy, Dose, and Mode of Administration: Participants in the SBR groups of Phase 2 and 3 will continue on their current complex ARV drug regimen.
Statistical Methods:
The analyses of this study will be structured in 2 portions: exploratory (Phase 2) to select the appropriate doses of the individual components of BIC and LEN as part of the BIC/LEN FDC, and confirmatory (Phase 3) to evaluate the treatment effect of BIC/LEN FDC.
Unique participants will be enrolled in the Phase 2 and Phase 3 portions of the study, respectively. Data from the interim and final analyses of each phase of the study will be analyzed separately. The confirmatory analyses will only include data from participants randomized into the Phase 3 portion of the study; they will not include data from participants in the Phase 2 portion.
Results from Week 24 primary analysis of Phase 2 portion will be used in conjunction with the results of Phase 1 rBA Study GS-US-621-6292 to select the dose of the BIC/LEN FDC tablet to be used in Treatment Group 1 of the Phase 3 portion, and the Phase 2 and Phase 3 Extension Periods of the study.
Phase 2:
The primary efficacy endpoint is the proportion of participants with HIV-1 RNA
≥ 50 copies/mL at Week 24 as determined by the US FDA-defined snapshot algorithm. The 2-sided 95% CIs for the difference in proportion of participants with HIV-1 RNA
≥ 50 copies/mL at Week 24 between each of the BIC plus LEN groups and SBR group will be constructed based on an unconditional exact method using 2 invert 1-sided tests.
The proportion of participants with HIV-1 RNA < 50 copies/mL at Week 24 as determined by the US FDA-defined snapshot algorithm will be analyzed using the same methods as for the primary efficacy endpoint.
The changes from baseline in CD4 cell count at Week 24 will be summarized by treatment using descriptive statistics. The differences in changes from baseline in CD4 cell count between each of the BIC plus LEN groups and SBR group and the associated 95% CIs will be constructed using an analysis of covariance (ANCOVA) models, including baseline
CD4 cell count as a covariate and treatment (each of the BIC plus LEN groups versus SBR) as a fixed effect in the models.
The number and proportion of participants experiencing treatment-emergent AEs (by system organ class [SOC], high-level term [HLT; if applicable], and preferred term [PT]), AEs leading to discontinuation of study drugs, and treatment-emergent laboratory abnormalities will be summarized by treatment using descriptive statistics.
For the general PK analyses, the PK of BIC and LEN may be evaluated using descriptive statistics and/or population analysis approaches. For the intensive PK substudy,
plasma concentrations of BIC and LEN may be summarized by nominal sampling time using descriptive statistics. Also, PK parameters (AUC0-t, AUCtau, AUClast, AUCinf, %AUCexp, CL/F, t1/2, λz, Vz/F, Cmax, Tmax, Clast, Tlast, Ctau, and Ct, as appropriate) may be listed and summarized using descriptive statistics.
The target sample size is 125 participants, with 50 participants in each of the BIC plus
LEN groups and 25 participants on SBR. The sample size in the Phase 2 portion of this study is determined based on practical considerations and past experience with similar types of studies. The sample size will provide an assessment of safety, efficacy, and PK to inform dose selection of BIC and LEN for the FDC tablet to be used in Phase 3 portion of the study.
Phase 3:
The primary efficacy endpoint is the proportion of participants with HIV-1 RNA ≥ 50 copies/mL at Week 48 as determined by the US FDA-defined snapshot algorithm.
The primary analysis will consist of a noninferiority test of switching to BIC/LEN FDC versus SBR, with respect to the proportion of participants with HIV-1 RNA ≥ 50 copies/mL at
Week 48, as determined by the US FDA-defined snapshot algorithm. It will be concluded that BIC/LEN FDC is noninferior to SBR if the upper bound of the 2-sided 95% CI of the treatment difference (BIC/LEN FDC – SBR) in the proportion of participants with HIV-1 RNA ≥ 50 copies/mL is less than 4% (ie, a margin of 4% is applied to noninferiority assessment). The 2-sided 95% CIs will be constructed based on an unconditional exact method using 2 invert 1-sided tests.
The proportion of participants with HIV-1 RNA < 50 copies/mL at Week 48 as determined by the US FDA-defined snapshot algorithm will also be analyzed using the same methods as for the primary efficacy endpoint based on the Full Analysis Set. However, noninferiority will be assessed using a margin of 10%. It will be concluded that BIC/LEN FDC is noninferior to SBR if the lower bound of the 2-sided 95% CI of the difference between treatment groups (BIC/LEN FDC − SBR) in the response rate is greater than −10%.
The changes from baseline in CD4 cell count at Week 48 will be summarized by treatment using descriptive statistics. The difference between the 2 treatments groups and the associated 95% CI will be constructed using ANCOVA models, including baseline CD4 cell count as a covariate and treatment (BIC/LEN FDC versus SBR) as a fixed effect in the model.
The number and proportion of participants experiencing treatment-emergent AEs (by SOC, HLT [if applicable], and PT), AEs leading to discontinuation of study drugs, and treatment-emergent laboratory abnormalities will be summarized by treatment using descriptive statistics.
For the general PK analyses, PK of BIC and LEN may be evaluated using descriptive statistics and/or population analysis approaches. For the intensive PK substudy, plasma concentrations of BIC and LEN may be summarized by nominal sampling time using descriptive statistics. Also, PK parameters (AUC0-t, AUCtau, AUClast, CL/F, t1/2, λz, Vz/F, Cmax, Tmax, Clast, Tlast, Ctau, and Ct, as appropriate) may be listed and summarized using descriptive statistics.
Participant-reported outcome instrument assessments may be summarized using descriptive statistics.
A total of approximately 546 participants, randomized in a 2:1 fashion to the BIC/LEN FDC versus SBR, achieves at least 90% power to detect a noninferiority margin of 4% in difference in percentage of participants with HIV-1 RNA ≥ 50 copies/mL at Week 48 between the
2 treatment groups. For the sample size and power computation, it is assumed that both treatment groups have 2% of participants with virologic failure (HIV-1 RNA ≥ 50 copies/mL) at Week 48 (based on the historical Gilead Genvoya® and BVY studies), that a noninferiority margin is 4%, and that the significance level of the test is at a 1-sided 0.025 level.